Recent advances in transcriptome sequencing have enabled the discovery of thousands of long non-coding RNAs (lncRNAs) across many species. Though several lncRNAs have been shown to play important roles in diverse biological processes, the functions and mechanisms of most lncRNAs remain unknown. Two significant obstacles lie between transcriptome sequencing and functional characterization of lncRNAs: identifying truly non-coding genes from de novo reconstructed transcriptomes, and prioritizing the hundreds of resulting putative lncRNAs for downstream experimental interrogation.
Researchers from the Broad Institute and the University of Massachusetts Medical School have devloped slncky, a lncRNA discovery tool that produces a high-quality set of lncRNAs from RNA-sequencing data and further uses evolutionary constraint to prioritize lncRNAs that are likely to be functionally important. Their automated filtering pipeline is comparable to manual curation efforts and more sensitive than previously published computational approaches. Furthermore, they developed a sensitive alignment pipeline for aligning lncRNA loci and propose new evolutionary metrics relevant for analyzing sequence and transcript evolution. Their analysis reveals that evolutionary selection acts in several distinct patterns, and uncovers two notable classes of intergenic lncRNAs: one showing strong purifying selection on RNA sequence and another where constraint is restricted to the regulation but not the sequence of the transcript.
slncky sensitively filters lncRNAs from reconstructed RNA-Seq data
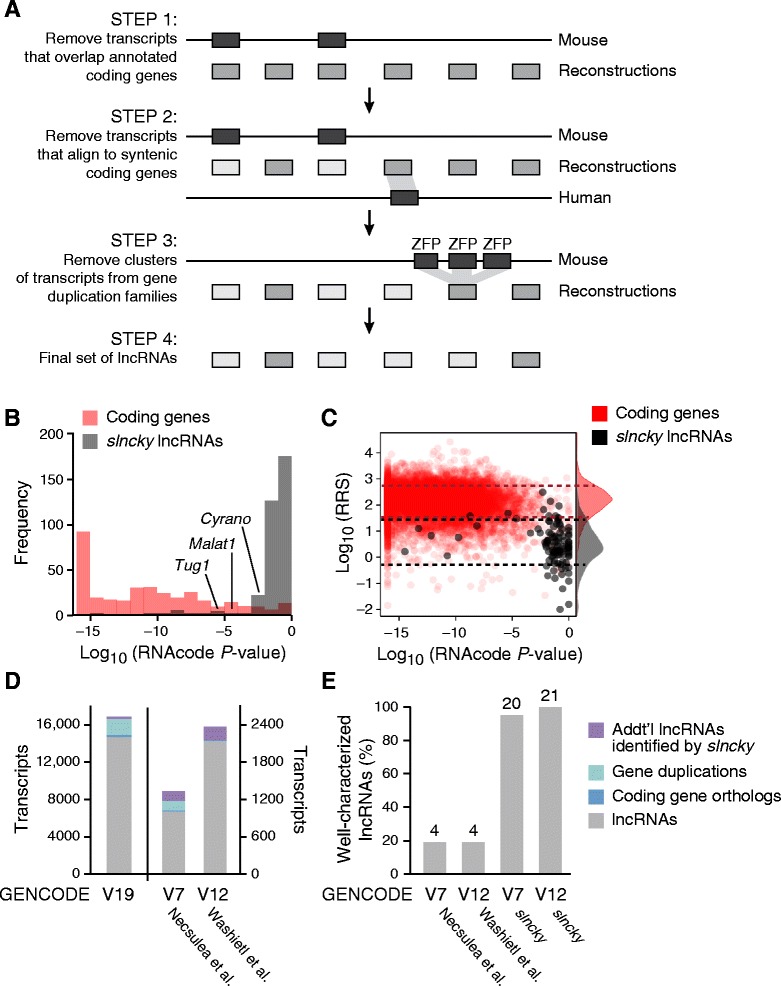
a Schematic of slncky’s filtering pipeline. Annotated coding genes are shown in dark gray, reconstructed transcripts in medium gray, and filtered transcripts in light gray. b Histogram of log10(P values) of coding potential as evaluated by RNACode for slncky-identified lncRNAs (gray) and coding genes (red). c Scatterplot of log10(P-values) of coding potential (x-axis) and log10(ribosomal-release scores) (y-axis) of slncky-identified lncRNAs (gray) and coding genes (red). Distributions of ribosomal-release scores (RRS) are displayed along right side of y-axis. Dotted lines denote one standard deviation above and below the mean of RRS distributions. slncky-identified lncRNAs have significantly higher coding potential P-values and lower RRS than coding genes. d Comparison of previously published sets of lncRNAs to slncky results. Number of transcripts also annotated as a lncRNA by slncky (gray), number removed by slncky as gene duplication or coding (light and dark blue), and number of additional transcripts annotated as a lncRNA by slncky but not the previous pipeline (purple). e Percentage of well-characterized lncRNAs identified in previously published sets compared to slncky results. Numbers above bars denote absolute number of lncRNAs
These results highlight that lncRNAs are not a homogenous class of molecules but rather a mixture of multiple functional classes with distinct biological mechanism and/or roles. The novel comparative methods for lncRNAs reveals 233 constrained lncRNAs out of tens of thousands of currently annotated transcripts.
Availability – The comparative methods for lncRNAs are available through the slncky Evolution Browser: https://scripts.mit.edu/~jjenny/